
- Services
- Industries
- Automotive
- Battery
- Building inspection
- Fire alarms system testing
- Household appliances
- Installation materials
- Industrial machinery
- IT & audio video
- Laboratory, test & measurement
- Lighting equipment
- Maritime, oil & gas
- Medical & healthcare equipment
- Military & aerospace product testing
- Wireless & telecom
- Resources
- About
- Blog
- Events
November 9, 2020
You have a new and innovative medical device that you believe can have a major impact on the health and wellbeing of millions of people. But to successfully bring your device to market, you must first navigate the seemingly complex process for the review and approval of medical devices in the European Union (EU), the U.S. and other major jurisdictions around the world.
And, if this is your first experience in seeking regulatory approval for a medical device, the path forward can certainly look overwhelming.
However, the process is not as complex as it might seem. To help clarify the specifics that you need to address, here’s a 10-step roadmap to help guide you through the regulatory review and approval process applicable to medical devices in most major markets.
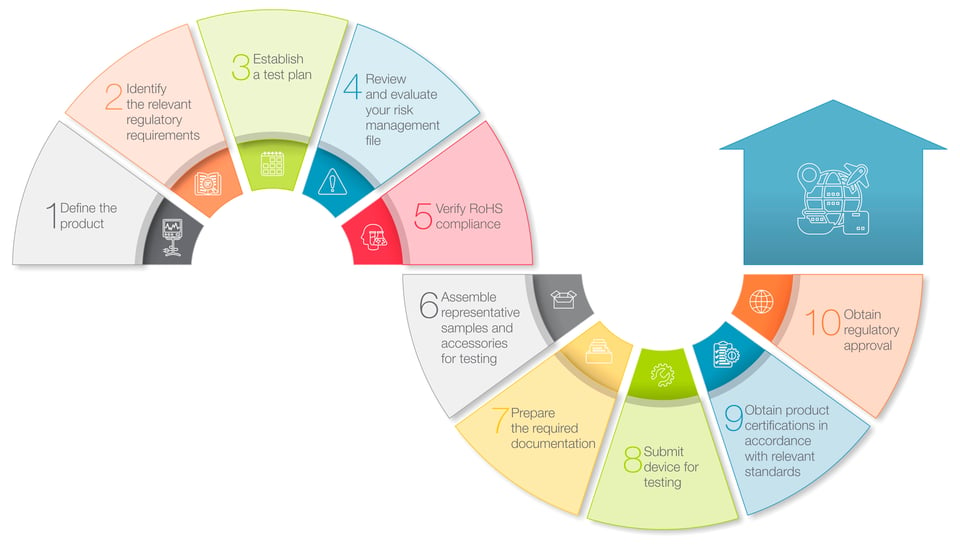
1. Define the product 
In this initial step, you work to confirm how and where your product is intended to be used. For example, medical devices used by trained medical professionals in established healthcare settings may be subject to different requirements than devices intended for use in home healthcare settings by consumers and caregivers. And, since regulations applicable to medical devices often differ from country to country, it’s also important to identify your intended target markets.2. Identify the relevant regulatory requirements
With step #1 completed, you now have the information you need to determine what regulations and standards will be used to evaluate the safety and efficacy of your medical devices. In addition to product-specific requirements, many regulators require that device manufacturers also provide documentation on their company’s quality management system (QMS), and may even require certification to a suitable QMS standard, such as ISO 13485.
3. Establish a test plan ![]()
With a detailed understanding of the requirements applicable to your device now in place, it’s time to develop a test plan that will generate the data necessary to demonstrate that your device is in compliance with the relevant regulations and standards. For devices that pose a higher degree of risk to patients, some regulators may require that you also conduct clinical trials to generate evidence supporting claims of product safety or efficacy.
An effective risk management strategy is a critical element in the development, production and post-market surveillance of medical devices. Compliance with the requirements of ISO 14971 provides an effective framework for identifying and evaluating all of the potential safety risks associated with a given product, and implementing the systems and controls necessary to reduce risk to acceptable levels.
5. Verify RoHS compliance ![]()
In certain jurisdictions, medical devices are subject to restrictions on the use of chemicals and other substances deemed to be hazardous. The EU’s RoHS (restriction of hazardous substances) Directive, for example, expressly limits the use of 10 banned substances in medical devices, including lead, mercury, cadmium and several different phthalates, and further requires device developers to prepare documentation that establishes compliance with the relevant requirements.
6. Assemble representative samples and accessories for testing ![]()
The next step in the regulatory review and approval process involves selecting and assembling representative samples of your device and any accessories that will be subjected to testing. Sample devices and accessories should mimic as closely as possible the final product design, so that they can be thoroughly evaluated for their functionality and safety in the anticipated use environment. And make sure that you’ve prepared enough samples to account for device failures and malfunctions that can occur during testing.
7. Prepare the required documentation ![]()
Now that you’ve completed steps #1-6, it’s time to prepare the documentation you’ll need to submit with your samples to the testing laboratory and to regulatory authorities. At a minimum, this includes a technical file or design dossier for your device, clinical evaluation reports, risk management files, RoHS-related documentation, and reports of any testing that you may have already conducted. Other documentation may be required, depending on the device’s risk classification or market-specific regulatory requirements.
8. Submit device for testing ![]()
Testing of medical devices can encompass a number of different aspects, such as performance, device safety and material biocompatibility. Depending on the design, features or functionality of a given device, testing might also evaluate electromagnetic compatibility (EMC) issues, such as radiated or conducted emissions, wireless interconnectivity, and potential risks associated with exposure to electrostatic discharge (ESD). Therefore, it’s important to select a testing laboratory that offers the full range of testing capabilities required for your device.
9. Obtain product certifications in accordance with relevant standards ![]()
The reports issued by your testing laboratory that document the results of testing should now serve as evidence that your medical device is in compliance with the relevant standards and is eligible for product certification. In some instances, self-certification such as a declaration of conformity (DoC) issued by your company is sufficient is to meet certain regulatory requirements. But for some standards, regulators are likely to require that testing data be reviewed by an authorized certification body such as an EU Notified Body, that then issues the required certification.
10. Obtain regulatory approval ![]()
With the relevant product certifications in hand, you’ve now reached the final step in the approval process, that is, the approval of your device by individual regulatory authorities, allowing you to legally market your device in their respective markets. The rigor of this final review and approval process as well as the process specifics varies from jurisdiction to jurisdiction. But obtaining regulatory approval first in either the EU or the U.S. can often speed the approval process in other jurisdictions.

The one additional aspect that all medical device developers and manufacturers must account for is the amount of time required to complete all of the above steps and achieve regulatory approval for their devices.
From beginning to end, the process typically takes months to complete, and can stretch to a year or longer in certain cases.
But following the roadmap we’ve presented here can help to determine early on if your device meets the applicable requirements and ensure that you’ve assembled the required data and documentation to eliminate surprises and unanticipated delays during the testing and approval process.
For more information, please watch our on-demand video, which will give you an introduction to the regulatory requirements for medical devices. You can also contact us anytime.
Disclaimer
This article is for general informational purposes only, and does not represent consulting or professional advice. Whereas all content is correct to the best of
our knowledge at the time of publication, Nemko does not undertake any responsibility for the accuracy or completeness of the information provided, the assessments made or any consequences arising from use of the document. Please seek Nemko’s support before acting upon any of the content.