
- Services
- Industries
- Automotive
- Battery
- Building inspection
- Fire alarms system testing
- Household appliances
- Installation materials
- Industrial machinery
- IT & audio video
- Laboratory, test & measurement
- Lighting equipment
- Maritime, oil & gas
- Medical & healthcare equipment
- Military & aerospace product testing
- Wireless & telecom
- Resources
- About
- Blog
- Events
February 10, 2021
As we’ve written about in a previous blog posting, documentation is a critical element in the regulatory review and approval process for medical devices. Yet, product development teams often fail to understand regulatory requirements regarding documentation that are applicable to their devices, or to devote sufficient time to preparing detailed documentation covering key aspects of their product development and testing processes.
Specific documentation requirements can vary, depending on a device’s risk classification or on market-specific regulatory requirements. But there are several documentation components that are commonly applicable to all types of devices and required by the majority of regulators.
Here’s a quick checklist that you can follow in assembling your documentation package prior to regulatory review and approval:
1. Technical documentation
This includes system drawings and illustrations, device schematics, printed circuit board (PCB) layouts, and wiring and insulation diagrams. It should also include a detailed bill of device materials, a list of critical components, and applicable datasheets.
In our experience, one of the documents that many customers have challenges obtaining is the insulation diagram. An example of the document can be found below.
For a pdf version, please click on the image.

Another document which many customers want to understand better is the list over critical components. An example can be found here.
2. Test reports and component certifications
Reports of preliminary testing conducted either by your organization or a trusted third-party provide documented evidence of your efforts to rigorously evaluate the safety and performance of your device. Component approvals (certificates and/or test reports) can significantly streamline device review and certification.
An example of a CB certificate can be found here.
3. Risk management file and supporting documentation
A risk management file prepared in accordance with the requirements of ISO 14971 and covering the risk management requirements in the IEC 60601 series provides details on your analysis, evaluation and control of all foreseeable risks associated with your device, and is mandatory for medical device approval in most markets.
Our experience is that a risk management file is often what result in delays since many companies don't know where to start. An example of a risk management file checklist can be found here. Many companies will ask design companies and consultants for assistance in this phase.
4. Software/PEMS documentation
ISO 62304 requires documentation of the development process for software that is integral to a given medical device, from planning and design through to implementation, verification and integration testing. In addition, the amended 3rd edition of IEC 60601-1 requires documentation of any programmable element (also referred to as “programmable electrical medical systems” or PEMS) integrated into a medical device.
5. Usability documentation
The documentation portfolio for a medical device should also include evidence of device usability, as defined in IEC 60601-1-6 and IEC 63266-1. Also known as human factors engineering, usability engineering evaluates potential risk associated with a device when used in accordance with prescribed procedures.
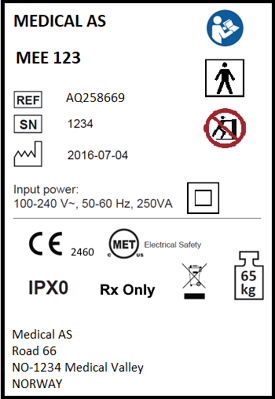
6. Marking labels
Marking labels applied or attached to a medical device provide important information to users, including warnings and other cautions. Descriptions and illustrations of all markings, symbols and signs should be thoroughly documented.
An example of some marking labels are seen below.
7. User instructions and other documentation
Finally, user instructions and all other documentation supplied with a medical device should be documented and cataloged.
Use this checklist to evaluate your own documentation collection and preparation processes. Better yet, find ways to integrate rigorous documentation requirements throughout the span of your organization’s product development cycle. Doing so can help avoid unexpected surprises and costly delays in successfully bringing your medical device to market.
Disclaimer
This article is for general informational purposes only, and does not represent consulting or professional advice. Whereas all content is correct to the best of
our knowledge at the time of publication, Nemko does not undertake any responsibility for the accuracy or completeness of the information provided, the assessments made or any consequences arising from use of the document. Please seek Nemko’s support before acting upon any of the content.